DeMixT Overview
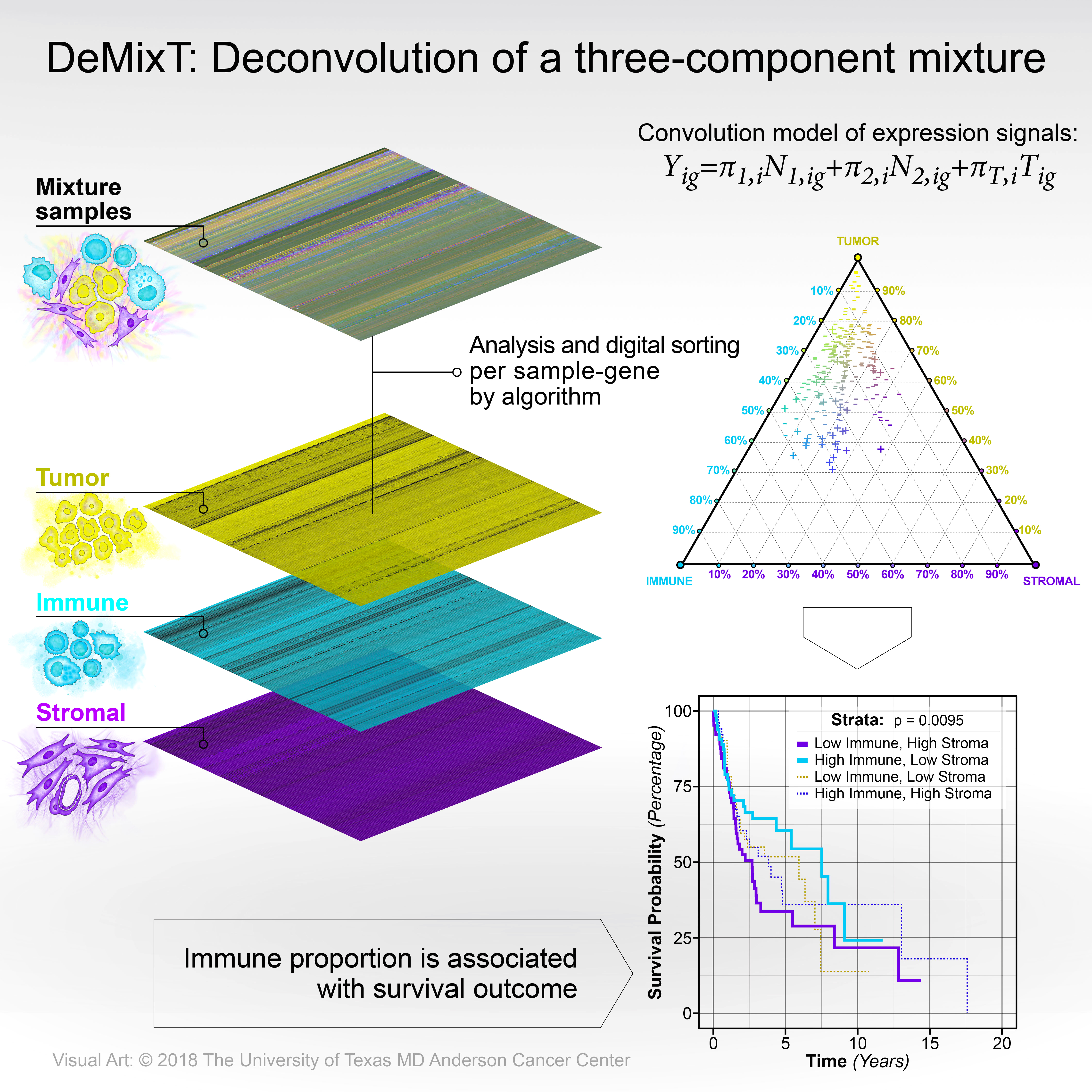
Transcriptomic deconvolution in cancer and other heterogeneous tissues remains challenging. Available methods lack the ability to estimate both component-specific proportions and expression profiles for individual samples. We present DeMixT, a new tool to deconvolve high dimensional data from mixtures of two or three cellular components (i.e. within heterogenous tissues such as cancers). DeMixT implements an iterated conditional mode algorithm and a gene-set-based component merging approach to improve accuracy. In a series of experimental validation studies and application across large datasets of cancer studies, DeMixT showed high accuracy in inference of cell-type specific proportions[1-2]. Improved deconvolution is an important step towards linking tumor transcriptomic data with phenotypes and clinical outcomes.
A tutorial for running DeMixT can be found here.
Reference
[1] Ahn, J. et al. DeMix: Deconvolution for mixed cancer transcriptomes using raw measured data. Bioinformatics 29, 1865–1871 (2013).
[2] Wang, Z. et al. Transcriptome Deconvolution of Heterogeneous Tumor Samples with Immune Infiltration. iScience 9, 451–460 (2018).
[3] Cao, S. et al. Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression. Nature Biotechnology Published online June 13 2022. doi 10.1038/s41587-022-01342-x.